[ad_1]
Viral DNA extraction
In previous studies, the range of yields of virome DNA extraction methods were low, typically below 50 ng per gram of faecal sample14. Thus, selecting a simple yet effective method is crucial for better characterizing the human gut virome. In this study the method selected for optimization was a method that utilized PEG precipitation, herein referred to as PEG-method, based on the report by Shkoporov and colleagues17. Briefly, homogenized faecal samples in buffer were filtered twice through 0.45 μm filters, followed by precipitation of viral particles with PEG. Viral particles were treated with DNase and RNase and lysed with proteinase K. Lysates were extracted with Phenol:Chloroform:Isoamyl Alcohol (PCIA) and purified using the DNeasy Blood and Tissue Kit following the manufacturer’s protocol. This method was selected due to its simplicity, cost efficiency, and low levels of bias with the aim of producing sufficient viral matter (estimated to be > 100 ng per gram) of faecal matter for long-read sequencing.
Initial extractions were carried out on a mock virome, consisting of Escherichia virus T4, Escherichia virus Lambda and Escherichia virus T3 phage. After consistently achieving 600 ng of DNA per gram of the mock virome, the method was applied on human faecal samples. DNA yield increased to an average of about 350 ng of DNA per gram of faecal sample in 60 μl of elution buffer (approx. 20 ng/μl) (see Supplementary Fig. S1 online), through a series of optimizations. These included increasing DNase and RNase concentrations to 10 U per sample each and incubation times to 2 h. Additionally, proteinase K incubation time was increased to 2 h and a step pooling three sets of 3 g of faecal material per sample from an individual was introduced. Thus, we established an amplification-free efficient virome DNA extraction protocol by optimizing an existing method with PEG precipitation (see Supplementary Fig. S2 online). The effectiveness of PEG-method supports a recent study that came to the same conclusion, suggesting that PEG is very suitable for use for virome DNA extractions in terms of cost and efficiency, providing relatively high yields of DNA and good purity of DNA compared to other methods18.
Viral prediction
Long-read sequencing was performed on faecal samples from six Orang Asli females (three Jakun and three Jehai). The number of reads from each sample varied greatly (Jakun: 20,118–289,003, Jehai: 47,072–375,911), which consequently affected number of contigs assembled by the program metaFlye19 (Jakun: 3–187, Jehai: 13–766). The average N50 (defined so that half the total nucleotides of an assembly belong to contigs of this length or longer) was 28,724 bp for the Jakun and 47,727 bp for the Jehai samples. The program VirSorter20 was used to predict viral contigs from six long-read sequences. In total, 61% (Jakun: 59%, Jehai: 62%) of microbial sequences were assigned as viral through VirSorter, suggesting that more than half the sequences were viruses. Additionally, when aligning these sequences to bacterial marker genes (using the program ViromeQC21) and human reference genome (version GRCh38), percentage of alignment was 0.54% (Jakun: 0.60%, Jehai: 0.48%) and 0.08% respectively. The remaining ~ 38% of sequences may be attributed to viruses of unknown bacterial or eukaryotic origin, suggesting these could lie within the viral dark matter. Viral dark matter comprises viral sequences that have not yet been identified, or are unknown contaminants. In previous virome studies such as human gut virome and ocean water virome, the amount of viral dark matter ranged from 40 to 90%22. Therefore, the amount of viral dark matter in this study is relatively low, but within expectations.
Jakun and Jehai Orang Asli viromes
The virome from individuals belonging to a semi-urbanised Jakun Orang Asli and a hunter-gatherer Jehai Orang Asli tribe from Peninsular Malaysia were characterized in this study. The analysis utilized metaFlye19 assembler program, followed by VirSorter20 virus identification, then the sequences were analysed in parallel with both the vConTACT223 taxonomic assignment program and the Genome Detective Virus Tool (GDVT)24.
Viral taxonomic assignments were assigned to all samples using vConTACT2 and plotted as a relative abundance plot (Fig. 1a). It was apparent that three viral families, Myoviridae, Podoviridae and Siphoviridae, dominated the virome. All three viral families originate from the same viral order, Caudovirales. Surprisingly, only Jehai samples were found to contain the viral family Myoviridae. Besides that, the abundances were not highly distinguishable between Jakun and Jehai.
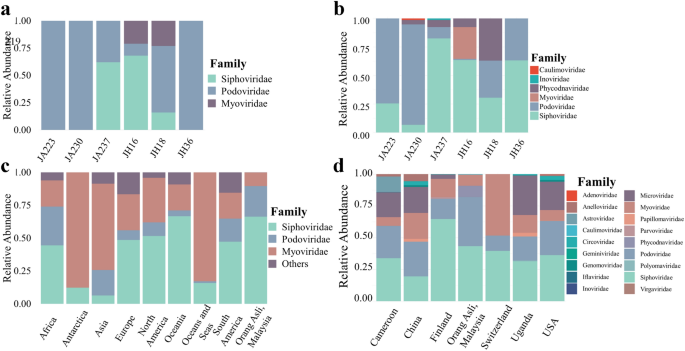
Relative abundance plot of viral families. (a,b) Six individuals, three from Jakun Orang Asli (JA223, JA230, JA237) and three from Jehai Orang Asli (JH16, JH18, JH36), taxonomy assigned by vConTACT2 and GDVT respectively. (c) Caudovirales order among Orang Asli, Malaysia, (n = 6) against different continents in isolates from NCBI Virus Database, Africa (n = 484), Antartica (n = 73), Asia (n = 8322), Europe (n = 6815), North America (n = 33,966), Oceania (n = 241), Oceans and Seas (n = 1052), South America (n = 161). (d) Orang Asli from Malaysia against Human Gut Virome Database separated based on country of origin, Cameroon (n = 29), China (n = 44), Finland (n = 56), Orang Asli (n = 6), Switzerland (n = 8), Uganda (n = 65), and USA (n = 279).
GDVT recovered a more diverse set of viral families (Fig. 1b). Consistently, the viromes of all individuals were dominated by the Caudovirales order, namely Myoviridae, Podoviridae and Siphoviridae viral families. However, all samples appear to have an increased assignment of Siphoviridae viral family with GDVT, perhaps due to the different database used as reference. Besides, the Myoviridae viral family which was found only in Jehai with vConTACT2, was also observed at low abundance in the Jakun with GDVT. The difference in the two tools may be attributed to the difference in database and techniques used to assign taxonomy, for example, some viruses present in one database that are not present in the other may have cause dissimilarities in the findings.
Interestingly, two samples had sequences attributed to plant viruses from the Caulimoviridae and Phycodnaviridae family. Phycodnaviridae has previously been reported in the infant gut virome25 and human oropharyngeal virome26, suggesting that it is highly likely that plant viruses can occasionally exist within the human gut depending on the diet of individuals. Phycodnaviridae have been reported to replicate within cells such as Chlorella27 A recent study stated that although Jehai Orang Asli do buy food, their purchasing power is low due to their socioeconomic status28.Therefore, they also rely on traditional food seeking practices to meet their dietary requirements. Interestingly, the same individual with the Phycodnaviridae viral family was predicted to have a large proportion of Tubulinea, which are a group of eukaryotic organisms including amoeba (see Fig. 4 later), suggesting potential amoeboid infection. The presence of Phycodnaviridae and Tubulinea in this individual may reflect the use of contaminated water sources and unsanitary living conditions and hence the origin of Phycodnaviridae viruses in these individuals may likely be from consumption of contaminated food or water.
Additionally, we observed the viral family of Inoviridae in one sample. This is a single stranded DNA (ssDNA) bacterial virus which would not have been detected through conventional short-read sequencing. For ssDNA viruses to be detected by short-read sequencing an amplification step is required, which may introduce amplification bias. The practicality of long-read sequencing to identify ssDNA viruses has also recently been demonstrated by Ji and colleagues29, as MS2 (ssDNA of the family Leviviridae) and PhiX174 (ssDNA of the family Microviridae) were both successfully classified with long-read sequencing.
Taxonomic assignments with both vConTACT2 and GDVT show that the most abundant viral order in the Orang asli gut virome is Caudovirales, which includes families Podoviridae, Siphoviridae, Myoviridae among others. This result supports previous virome findings that most sequences consist of the order Caudovirales9. In addition, the presence and abundance of viral sequences were different across all six samples, suggesting a high interpersonal variability in the human gut viromes of these indigenous populations (Fig. 1b).
The noteworthy difference between the Orang Asli groups was the richness of viral family Podoviridae and the scarcity of viral family Myoviridae in the Jakun compared to the Jehai (see Supplementary Fig. S3 online). However, it should be noted that the small sample size hinders any statistical significance from being assigned to these differences in the gut viromes of the two communities. Despite that, these findings are suggestive evidence that there might be virome differences between the two populations.
During finalization of this manuscript, we became aware of Luis et al.30 who described a new viral database, the Gut Phage Database and their finding that most of the viruses belong to p-crAssphage which has been reported to possess a morphology similar to Podoviridae family31. The applicability of their findings to the Orang Asli we have described require further investigation.
Malaysian Orang Asli and worldwide viromes
Considering the high levels of viral order Caudovirales among the Orang Asli population in this study, the taxonomy of the Orang Asli virome was compared with viral families from the viral order Caudovirales deposited in the NCBI Virus Nucleotide Database (Fig. 1c) as of 17th January 2020 and stratified by the continent of origin. Viral families Siphoviridae, Podoviridae, and Myoviridae dominated regardless of the continent of origin. However, the abundance of each viral family varied drastically depending on continent, suggesting a possible role of geographical effects on dissimilar abundance of viral families.
Additionally, the taxonomy of the Orang Asli viromes (this study) assigned by GDVT were also compared to the Human Gut Virome Database (HGVD) (Fig. 1d), which comprises metagenomic human gut virome studies compiled from several countries. Surprisingly, the Orang Asli groups did not contain any of the ssDNA viral family Microviridae, which is in considerable abundance in several countries, most notably Cameroon (20%), China (20%), Uganda (30%), and USA (22%) (Fig. 1d). This may indicate that Microviridae could be less prevalent in indigenous Malaysian populations compared to other populations. However, previous studies utilized multiple displacement amplification (MDA) that preferentially amplifies ssDNA, suggesting a possible bias in frequency of ssDNA from metagenomics studies32. The family Phycodnaviridae which was found in this study, was absent in the HGVD, suggesting that it may be region specific and hence, perhaps explains not identifying it when using vConTACT2 to characterize the viral sequences.
Alpha diversity measures the number of species within a given area or ecosystem. The alpha diversity measures used here are estimators (Chao1 and Inverse Simpson index) of diversity based on the abundance and number of species. Despite the variations in viral families, there was no significant difference in alpha-diversity between any countries compared here (Fig. 2a,b; see Supplementary Table S1 online), suggesting that virome richness remains consistent irrespective of geographical region. Beta diversity describes how much of the total diversity observed within a group or set is due to a particular member of a set. Thus Fig. 2c,d display the beta-diversity of the named country relative to the total diversity of the set composed of the named country and the Orang Asli. The further the named country samples are from 0,0 the more different the viruses are in that country are from the ones found in the Orang Asli. Beta-diversities between all the countries and the Orang Asli samples were tested using permutational multivariate analyses of variance (PERMANOVA) using Bray–Curtis distance matrices (Fig. 2c,d) and having a significance value of p < 0.001 in all cases (see Supplementary Figs. S4 and S5 online). It was found that the virome beta-diversity of Orang Asli, Malaysia group from this study was significantly different to all other countries in HGVD, indicating that the Orang Asli, Malaysia virome compositions were highly dissimilar compared to virome compositions identified from other countries. However, after carrying out analysis of the dispersion of the samples, a possible confounder of the geographical effects found here, it was seen that the Switzerland samples had a significantly different dispersion to all other samples and so differences observed in PERMANOVA with the Switzerland samples cannot be relied on (also USA-Cameroon; Supplementary Fig. S11 online).
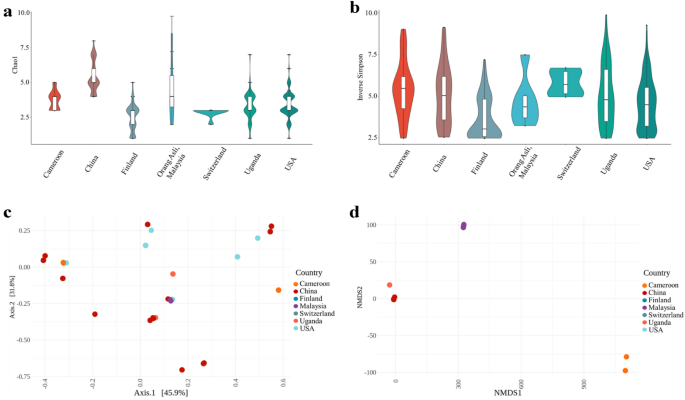
Diversity metrics for Orang Asli from Malaysia and worldwide metagenomics virome data from HGVD. (a,b) Viral alpha diversity. The boxplots show the alpha diversity of the viral communities in various countries compared to Orang Asli from Malaysia by means of viral sequences using alpha-diversity indexes; (a) Chao1 and (b) Inverse Simpson. (c,d) Viral beta diversity. The two-dimensional scatter plot shows the beta diversity of viral compositions in various countries, Cameroon (n = 29), China (n = 44), Finland (n = 56), Switzerland (n = 8), Uganda (n = 65), and USA (n = 279) compared to Orang Asli, Malaysia, (n = 6) generated by ordination methods; (c) principal coordinate analysis (PCoA) and (d) Non-metric multidimensional scaling (NMDS) from Bray–Curtis distance matrix.
Putative viral hosts
Putative hosts for viral sequences were determined by GDVT (Fig. 4 and Supplementary Fig. S6 online) with 67% of viruses assigned a potential bacterial or eukaryotic host. The abundance of viral host was approximated by the abundance of each virus. In total 33% of the viruses had an unknown host. The most abundant putative host among all samples were from the Firmicutes phylum, with 60% of Jakun Orang Asli and 28% of Jehai Orang Asli viromes being assigned to this phylum. Additionally, Bacteriodetes and Proteobacteria hosts are also deduced to contribute 4% and 6%, respectively, of the gut viromes in the Orang Asli.
Functional profile of Orang Asli viromes
To functionally profile the gut DNA virome, viral sequences were annotated using the program DRAM33. Open reading frames were screened to determine viral genes and assigned classifications, summarized in Table 1 as either viral hypothetical genes, viral genes with unknown function, viral genes with host benefits, viral genes with viral benefits, viral replication genes, or viral structure genes based on VOGDB functional classification (https://vogdb.org).
Predictably, the lack of knowledge on the virome contributed to the large proportion of hypothetical genes, amounting to 91% and 94% among Jakun individuals (n = 1598) and Jehai individuals (n = 11,849) respectively. Furthermore, the category of viral genes with unknown functions yielded 105 genes (5.99%) in Jakun individuals and 465 genes (3.70%) in Jehai individuals. This proportion of hypothetical and unknown genes demonstrates the need for a thorough investigation on the human gut virome, including the use of other omic approaches such as metatranscriptomics, metaproteomics and metabolomics to gain a better grasp of unknown and hypothetical genetic factors of microbes as well as the human host. Conversely, 27 genes (1.54%) in Jakun and 204 genes (1.62%) in Jehai viromes were designated as viral genes with host benefits, which are important for the evolution and persistence of the host. For instance, the gene anaerobic ribonucleoside-triphosphate reductase (nrdD) was found in a Myoviridae virus, which is responsible for the catalysis of ribonucleotides into deoxyribonucleotides, essential for DNA synthesis and repair in bacteria. Similarly, Integrase genes (int) are important for gut microbiome homeostasis because they allow viruses to transfer genes to bacteria which may enhance bacterial metabolic functions, eliminate competing organisms, or increase virulence34. For example, some viruses enhance bacterial metabolic functions by having the ability to promote biofilm formation in bacteria, subsequently improving bacteria fitness35.
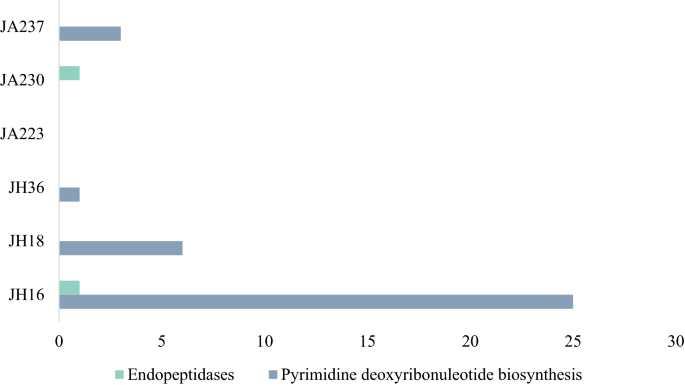
Viral genomes often also acquire host metabolic genes, known as auxiliary metabolic genes (AMG), which could improve viral fitness and promote viral reproduction. AMGs were identified for all samples and are summarized in Supplementary Table S4 online. AMG modules found in more than two samples were Endopeptidases and Pyrimidine biosynthesis (Fig. 3). Endopeptidases act as an endolysin at the end of an infection cycle36, hence the detection suggests a presence of lytic bacteriophages in the human gut. The presence of pyrimidine biosynthesis has previously been reported in marine viruses and is found to be highly important in viral replication37. Additionally, host cells (bacteria and eukaryotic cells) may need to utilize pyrimidine biosynthesis pathways from viruses to supply sufficient nucleoside 5′-triphosphate (NTP) for fast-dividing cells such as the intestinal epithelial cell lining the human gut and fast-replicating viruses38. AMGs involved in the pyrimidine pathway that are found in this study are dUTP pyrophosphatase (dUTPase) and ribonucleoside reductase (RNR). dUTPase prevents misincorporation of uracil39 and is crucial in maintaining accurate viral genome replication and has been found in many viral genomes such as Epstein-Barr virus40, Feline immunodeficiency virus41, and African swine fever virus42. RNR is responsible for changing the ribonucleotides into deoxyribonucleotides, which are necessary for DNA synthesis43. Interestingly however, Enav and colleagues37 found that both purine and pyrimidine biosynthesis pathways were enriched in the AMGs, whereas in this study only pyrimidine biosynthesis were annotated (Fig. 4).
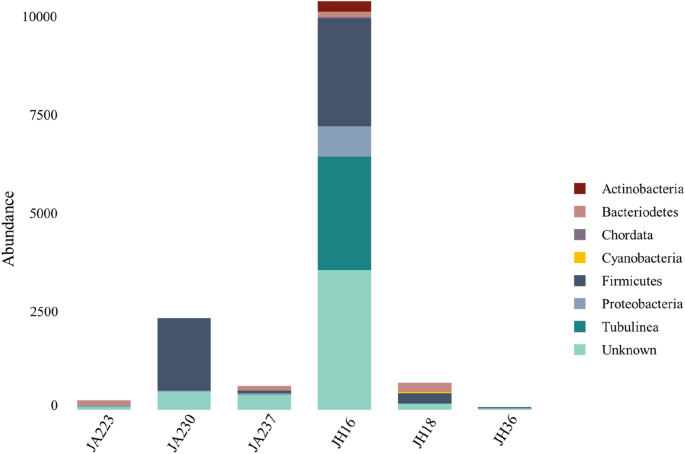
Abundance of putative hosts in six samples. Three individuals from Jakun Orang Asli (JA223, JA230, JA237) and three individuals from Jehai Orang Asli (JH16, JH18, JH36) taxonomy assigned from GDVT, expressed as counts of putative bacteria depending on viral abundance.
Auxiliary Metabolic Genes in the Virome of six Orang Asli. Three individuals from Jakun Orang Asli (JA223, JA230, JA237) and three individuals from Jehai Orang Asli (JH16, JH18, JH36).
Sequencing platform validation
We compared two sequencing platforms comprising of the conventional short-read Illumina sequencing and third generation Oxford Nanopore Technology (ONT) long-read sequencing. Two samples were chosen (one from each Orang Asli group) labelled as JH16 (Jehai) and JA230 (Jakun) for sequencing on both platforms. By analysing sequencing depth against the number of assembled contigs, it is apparent that at 800 MB of Oxford Nanopore long-read or 20 GB of Illumina data per sample, the virome is not fully captured (see Supplementary Fig. S7 online). However, long-read sequencing readily assembles close to complete viral genomes as it was able to achieve greater N50 values and percentage of viral sequences in both samples compared to short-read sequencing (see Supplementary Fig. S8 and Table S5 online). This is likely due to short-read sequencing capturing only fragments of a genome in a single read, which often cannot be attributed to any specific viral genome. Long-read sequencing however, could theoretically cover, for many of the viruses, an entire genome in a single read. Hence, the low sequencing depth of long-read sequencing is overcome by the improved coverage of individual viral sequences and so it appears that breadth of coverage outperforms depth of coverage in this study.
Viral taxonomic annotations performed using vConTACT2 (see Supplementary Fig. S10 online), found that short-read sequencing revealed greater viral diversity compared to long-read sequencing. However, the low sequence coverage of viral sequences may also be involved in skewed misassignments of viral annotation in short-read sequencing. In summary, currently both sequencing technologies should be used in combination to provide a greater perspective of the virome, as short-read technologies provide greater sequencing depth while long-read provides closer to complete viral genomes. Recent improvements in long-read sequencing, such as High-Fidelity sequencing on PacBio Sequel II Systems, may provide alternate methods for investigating viromes in any environment. However, this may often not be possible due to resource constraints.
[ad_2]阅读更多